My major work:
HGTector: Genome-wide horizontal gene transfer discovery via BLAST hit distribution statistics.
And a list of other works:
SubDBer: Sequence database subsampler for metagenomic analysis.
All-Branch PAML: Batch selection test with specified model sets on every branch of a tree.
plotMLE: Assess quality of BEAST MLE model test by plotting result against # generations.
BeforePhylo: Manipulate multiple sequence alignments for phylogenetic reconstruction.
AfterPhylo: Manipulate trees after phylogenetic reconstruction.
ppt2doc: Convert PowerPoint slides into printer-friendly Word documents.
Folder Adorner: Memory of my first commercial software product (1999).
And a few handy scripts for trivial / non-trivial tasks.
*Note: Click title to download
*PS: I want to thank Dr. Katharina Dittmar for supporting me to combine my interest in programming and serious scientific research, without which I would not have had an intersection with a bioinformatician's career path.
HGTector
A novel BLAST-based approach for genome-wide surveys of putative HGT events, coded in Perl. Unlike conventional BLAST-based methods, which rely merely on the best match, this method features the statistical analysis of BLAST hit distribution patterns of genomes with phylogenetic consideration of the organisms of interest. Thus it is notably insensitive to stochastic effects that can lead to false positives and false negatives. The process is rapid, exhaustive, and computationally inexpensive.
Please check out our original publication that details this method. Also please find a poster we presented during GLBIO 2013:

Coded in Perl, and optionally calls R to perform statistical analyses. The program also comes with a GUI, written in HTML + Javascript. The whole package is available from GitHub.
Direct download links: version 0.2.0 (latest) or version 0.1.9.
SubDBer
Flexible taxonomy-based subsampling of sequence database for efficient and focused metagenomic analyses. Example:
python subDBer.py -in nt -out sub_nt -outfmt blast -within 2 -exclude 1117 -rank genus -size 1 -keep 816,838,1263
This command takes the NCBI nt database as input -> starts with all organisms from Bacteria (TaxID: 2) except for Cyanobacteria (1117) -> picks one representative organism per genus -> except for Bacteroides (816), Prevotella (838) and Ruminococcus (1263), in which all organisms are included -> creates a new BLAST database sub_nt that contains sequence data from the selected organisms.
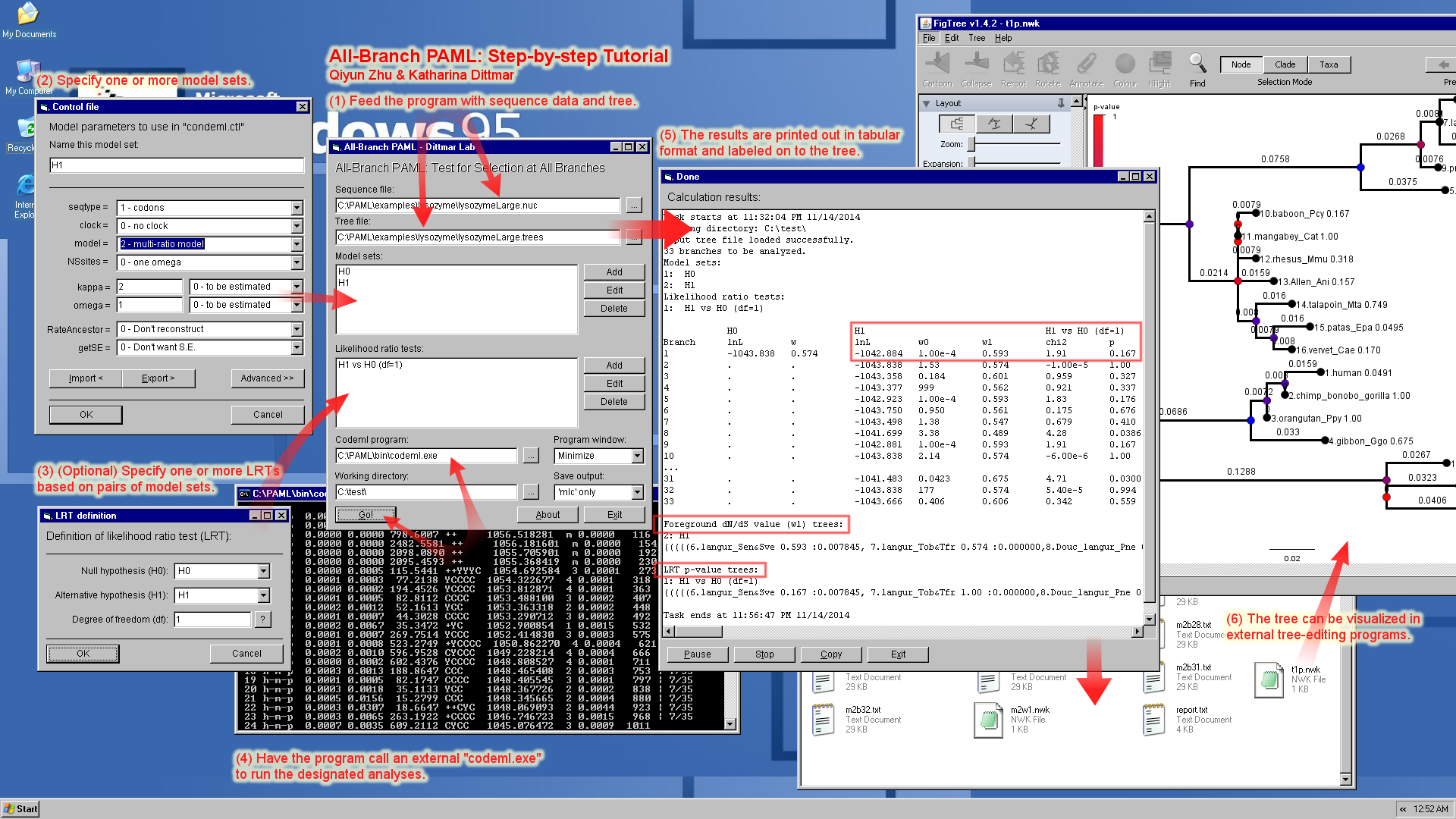
All-Branch PAML
An automated pipeline for selection test with designated model sets on each and every branch of a tree. Written in VB6.
This Program calls the PAML package to test selection on each and every branch of a phylogenetic tree. It runs the
codeml program with two (or more) alternative model sets (codeml.ctl),
and performs likelihood ratio test (LRT) to assess the significance of
the difference among likelihoods. The resulting p-values as well as the
foreground omega (dN/dS) values are reported in tabular form, and also
added to the tree as branch labels for further visualization. Here is a one-image screenshot + feature demonstration + tutorial:
plotMLE
Plot BEAST marginal likelihood estimation (MLE) results against number of generations, to assess whether the MCMC chain is lengthy enough.
MLE by PS or SS (Baele et al., 2012) is a statistically robust and computationally expensive approach to test alternative models in a Bayesian phylogenetic inference. It was considered to be superior to conventional Bayes Factor approaches. However, it could take very loooooong time on any advanced computer to get a reasonable (stable) MLE value.

To assess whether one has finally executed adequate MCMC generations in MLE, they can plot the MLE value against the number of generations, and see if the curve has entered a stable plateau. plotMLE.pl is the script that automates this process. Feed it with the .mle.log file generated in a regular BEAST run, and it will print out a table:

Which can be nicely plotted:

Above is an example, which tests HKY against TN93 models on the BEAST sample data "primates.nex". As you can see, after a ton of generations, the two curves are still creeping up and down, but the trend is clear that blue is higher than red.
The script is in Perl and relies on no dependency but the BEAST 1.8.x program.
Below are two scripts I created to ease my own phylogenetic analyses. I made them end-user ready and released to the public.
BeforePhylo
A Perl script for manipulating multiple sequence alignments for phylogenetic reconstruction. Features:
- Concatenate multiple MSAs and generate partition table for RAxML, MrBayes and BEAST
- Remove empty sites, fill gaps and/or ends, replace ambiguous codes with 'N's
- Translate taxon names into numbers or based on a dictionary
- Extract subset of sequences based on a list
- Divide MSA by codon position or by a partition table
- Convert formats (FASTA, NEXUS and Phylip)
Example:
perl BeforePhylo.pl -filter=subset.txt -N -trim -fillends -conc=mrbayes 16S.fas ftsZ.fas gltA.fas groEL.fas rpoB.fas
This command will take the five input MSAs, extract a subset of taxa defined in subset.txt, concantenate into a master matrix, remove empty sites, replace ambiguous codes and end gaps with "N", generate a MrBayes input file (.nex), which includes sequence data and partition definitions.
Available from GitHub.
AfterPhylo
A Perl script for manipulating trees after phylogenetic reconstruction. Features:
- Annotate tips (IDs -> full taxa names).
- Compute average confidence value across tree.
- Scale up/down branch lengths.
- Remove branch lengths and keep topology only.
- Collapse nodes with confidence values lower than a cutoff.
- Convert between Newick and Nexus formats, with confidence values kept.
- Compatible with trees generated by RAxML, PhyML, MrBayes, BEAST or other popular phylogenetics programs.
Example:
perl AfterPhylo.pl -confonly -collapse=50 -annotate=names.txt -replace -format=newick tree1.nex
This command will take a MrBayes output tree (tree1.nex), keep confidence values (posterior probabilities) only while getting rid of all other junks, collapse any node that receives a posterior probability lower than 50% (major rule), replace taxon names with a designated list of actual organism names (e.g., Dmel => Drosophila melanogaster), and export in Newick format.
Available from GitHub.
PPT2DOC
As the name suggests, it converts a PowerPoint presentation into a Word document. It is written in VB6 + Office VBA.
I wrote it in 2005 for my classmates, so that we can print out lecture notes in a more organized form. Recently, I was surprised to find that it still functions correctly in modern software environments (that is, Windows 8.1 + Office 2013). So I polished and post it again.
Here are the screenshots. Before (PowerPoint):

After (Word):

Folder Adorner
Dating back to 1999, this was my first software product formally released to the market (i.e., "shareware"). I sold a few copies of it. This program can add some interesting visual and functional features to a folder in Windows 98/2000 systems. The major secret is to edit the "desktop.ini" file. Written in VB5.
I was surprised to find that it is still available in the original purchase website. Though I honestly don't think it is still effective in modern Windows7/8 systems.
Here is a screenshot:

A few handy scripts for trivial / non-trivial tasks:
simEvol.cpp: A simple program that simulates nucleotide sequence evolution, from a given starting sequence, under a given mutation rate matrix, through a given phylogenetic tree.
It is a single-file C++ program and does not rely on any third-party dependencies.
Prot2DNA.py: A Python script to batch-retrieve DNA sequences from the NCBI server based on protein entries.
More to be added.
No comments:
Post a Comment